
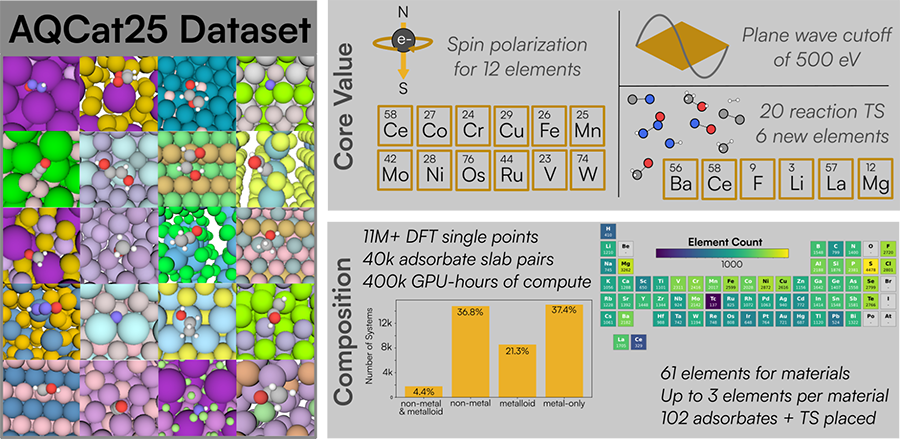
We’ve produced a dataset for AI catalyst design. The dataset, AQCat25, contains 11 million Density Functional Theory (DFT) calculations of energies from 40,000 unique catalytic systems. Compared to previous datasets such as OpenCatalyst 2020, AQCat25 has higher accuracy, covers six new elements and twenty reaction transition states, and includes spin polarizations for twelve elements. Using NVIDIA DGX cloud hardware, we created AQCat25 to help replace lab-based catalyst design with computation.
At SandboxAQ, we drive deep impact at scale. We do this via what we call Large Quantitative Models (LQMs) - AI models trained on scientific, rather than linguistic, data. In our most recent post about SAIR (Structurally Augmented IC50 Repository), we described a path to expand the available data for protein structure prediction, key to our drug discovery focus area. Here, we show work in another focus area, catalyst design and optimization.
A catalyst is a substance that increases the rate of a chemical reaction without being itself consumed (see Figure 2). Better catalysts can make new reactions effectively possible, more efficient, or less harmful. Since most industrial chemical processes somehow involve a catalyst, improvements to catalyst design improve the overall economy.
For example:
Figure 2 shows how catalysts make chemical reactions more efficient. On the left, we see a hypothetical chemical reaction: water being stripped of a hydrogen atom. Once stripped, the water has less energy, so the reaction is stable. To perform the stripping, energy must first be added to reach the transition state, for example by heating. Catalysts lower the energy of the transition state, so less needs to be added. Better catalysts lower it more.

To test potential catalysts, we need to determine the rate of reaction. Experimentally, we physically perform the reaction in the presence of different catalysts, which thus must be expensively synthesized and tested. Algorithms such as DFT can instead calculate the relevant energies and project the rate of reaction. Such computations can currently be more expensive than experimentation: for example, AQCat25 consumed 400,000+ GPU-hours using NVIDIA DGX H100 cards, but LQM models trained on their results are thousands of times faster.
AQCat25 provides a large and diverse collection of DFT relaxation trajectories for heterogeneous catalysis, and is intended primarily for training and testing machine learning interatomic potentials. It includes a wide range of adsorbates and materials, including both in-domain (ID) and out-of-distribution (OOD) splits for robust model evaluation. It makes three main improvements upon existing open source datasets:
You can learn more about the dataset organization on Hugging Face. Any split can be quickly loaded and filtered to find specific structures, without needing to download the full archive.
AQCat25 is free for non-commercial use under the CC BY-NC-SA 4.0 license. If you are interested in using AQCat25 for commercial applications, or if you have any other questions, requests or feedback, please email us at AQCat25@sandboxaq.com.