We’re releasing SAIR (Structurally Augmented IC50 Repository), a dataset of 5,244,285 computationally folded three-dimensional protein-drug molecule co-structures - 5 each per unique protein and drug molecule - tagged with experimental potency data. This dataset enables the creation of better models to predict structures and potencies, by selecting only structures which result in the best agreement between computational potency predictions and the experimental tags.
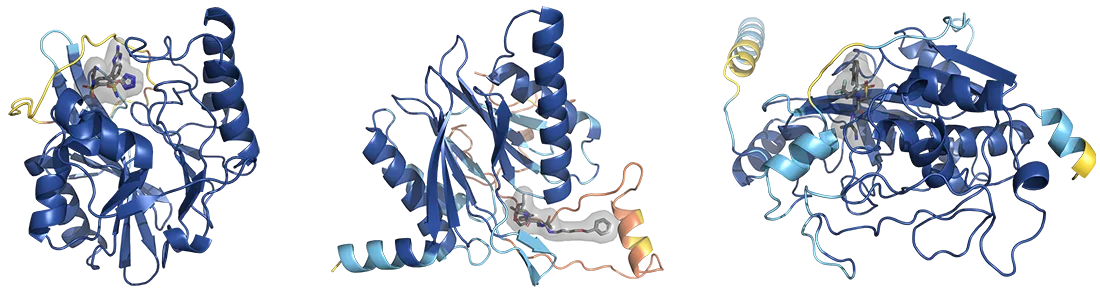
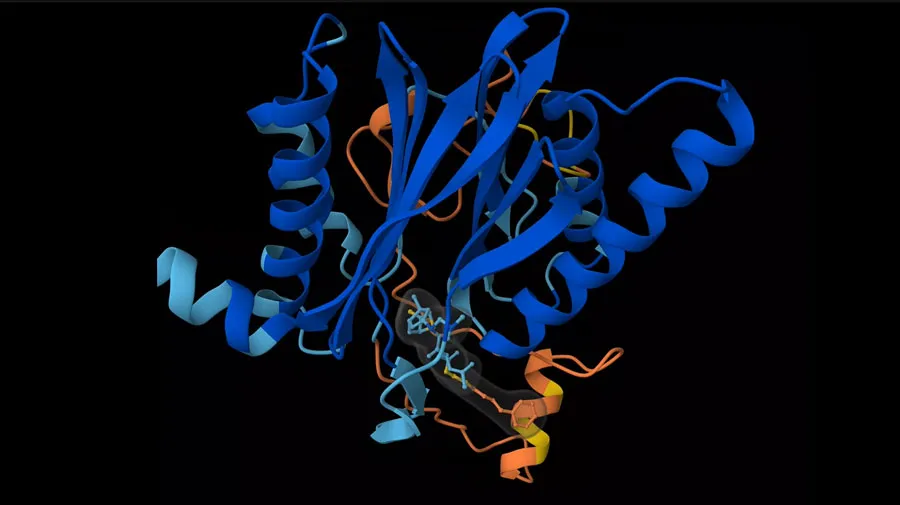
To get a feel for the SAIR dataset, let’s begin by taking a look at Figure 1, which depicts three entries. In this figure, the ribbon-like shapes represent proteins that can be found in the human body. The smaller structures highlighted by grey clouds are drug molecules, “bound” to the proteins. The SAIR dataset includes the predicted structures of the proteins, and the positions - or “poses” - of the molecules. It also includes experimental measurements of the so-called potency of the drug, which relates to how strongly the drug binds to its target protein. As the poses and potencies of new candidate drug molecules are critical information in drug discovery, we expect the SAIR dataset to facilitate the creation of new computational methods to identify new drugs.
Roughly speaking, early-stage, small-molecule drug discovery is a process of identifying new candidate molecules that strongly bind to a specific target protein, related to a disease of interest. To achieve this, computational chemists roughly use a workflow like the following:
The above process is then iterated and refined, until a molecule with enough potency is found. This molecule then becomes a drug candidate to treat the disease; and is then subject to further testing to assess its potential as a drug, a process called drug development, as opposed to drug discovery.
For quite some time, a major goal of computational drug discovery research has been to compress the above procedure into a single step, by training machine learning models to predict potencies directly from a specification of a protein (without its three-dimensional structure) and a molecule. Such models might even go so far as to invent new molecules from scratch, instead of scanning over libraries.
Giant steps have already been made. Models like AlphaFold and AlphaFold2 have been able to predict structures with impressive accuracy for some time - but not poses or potencies. Co-folding models such as AlphaFold3, Boltz-1, and others have more recently demonstrated a good ability to predict poses as well. Most recently, Boltz-2 has demonstrated promising results in directly predicting potencies. But broadly speaking, these models continue to struggle with their ability to make accurate enough predictions to be consistently useful in practice, especially when applied to proteins or molecules which are very different from those they were trained upon.
The obvious solution is to train on more data. Unfortunately, most of the publicly available protein structure data have already been used for training. Since the necessary experiments are slow and expensive - this is, after all, the basic problem co-folding models aim to solve - creating enough experimental data to change this is expensive and time consuming. We created SAIR to show a way around this.
While experimental structural data is slow and expensive to make, experimental potency data - direct measurements of how strongly a given molecule binds a given protein, without knowledge of the structure or of the pose - is much cheaper. Large databases, such as ChEMBL and BindingDB, exist cataloging such data.
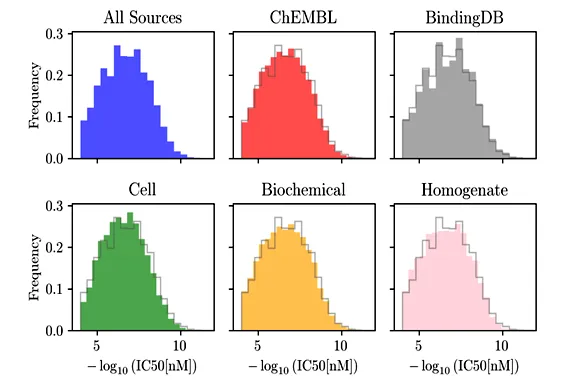
We created SAIR to leverage such data against structure and potency prediction. To do this, we used the Boltz-1x co-folding model to make multiple predictions of structures for each entry in the ChEMBL and BindingDB databases - see Figure 2 for histograms detailing the exact distributions of data we used. Because of randomness in the computational prediction, these predicted structures differ from one another, and the difference is greater where the model is least accurate. By comparing these redundant structures and potencies with an alternative, computational potency measurement, we can select the most accurate ones yielding the best predictions. SAIR thus enables dramatic improvement of both co-folding and potency prediction models.
Alongside the structures, we release several complementary pieces of information that may be useful to researchers when using this data. We include the results of running an algorithm called “PoseBusters”, which dictates if a generated pose is valid according to known laws of chemistry and physics. Only approximately three percent of our generated poses fail this test, which further validates the usefulness of SAIR.
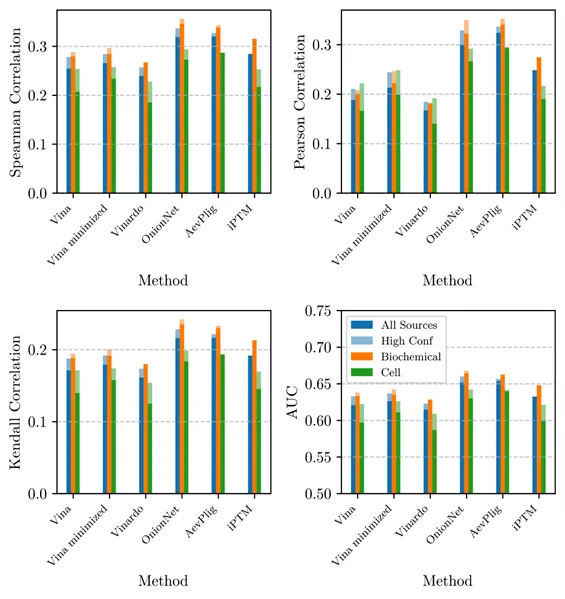
As an initial demonstration of how researchers might use SAIR, we performed a study of various different potency prediction methods as applied to dataset entries obtained from two different types of experiment, biochemical and cellular. Accurate methods should be expected to perfectly predict both types of experiment. Results are depicted in Figure 3; we find that the deep learning methods OnionNet and AevPilg exhibit the highest correlation. This is a promising validation of the machine learning based approach in general.
Our hope is that researchers will use SAIR to advance the accuracy and power of computational drug discovery. We are especially excited by the prospect of creating inexpensive and accurate new predictions for proteins which might be difficult to image experimentally, especially “dark” proteins that may not be direct drug targets, but might nevertheless be critical to understanding the behaviour of cells in response to a drug. More details about the data and our affinity experiments can be found in our manuscript.
We’re releasing SAIR as free and publicly available data under the Creative Commons Attribution License (CC BY 4.0). The data are completely free for commercial and non-commercial use.
Please visit us at www.sandboxaq.com/sair to learn more and download the dataset.