SandboxAQ has launched a GPCR Virtual Screening Solution that helps drug developers predict both which compounds bind a receptor and whether those compounds will activate it or block it; two questions that together determine whether a GPCR drug produces the intended therapeutic effect.
The platform, AQState, predicts both binding affinity and mechanism of action before experimental testing begins. For teams running GPCR programs, this can mean a more focused candidate list and less reliance on costly follow-on screening to determine pharmacology downstream.
GPCRs are signaling proteins on the surfaces of cells that act as molecular switches. Drugs that bind them can flip the switch in either direction. An agonist activates the receptor; an antagonist prevents activation. That distinction determines the drug's therapeutic effect. An error at this stage can reverse the intended effect entirely.
Standard virtual screening ranks compounds by predicted binding affinity: how tightly a molecule is likely to stick to a target. For GPCR programs that require a specific pharmacology, that leaves a critical question unanswered: will this compound activate the receptor or block it?
Without that answer, teams must run costly experimental assays or follow-on computational analyses to separate agonists from antagonists, steps that filter the hit list rather than improve it, at significant time and cost.
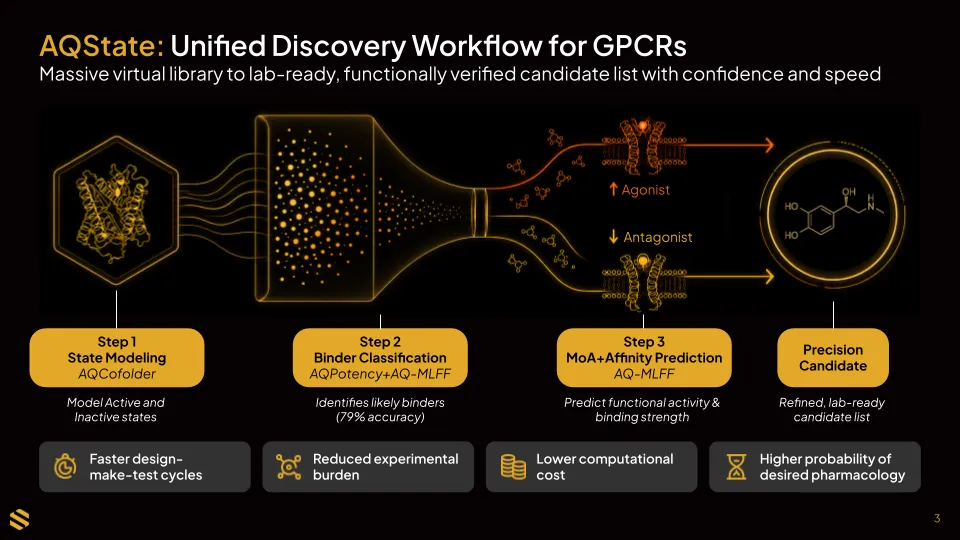
GPCR receptors can exist in two meaningfully different shapes: an active state that triggers a biological signal, and an inactive state that does not. Whether a drug acts as an agonist or antagonist depends on which state it preferentially engages. Predicting that requires accurate structural models of both. Many drug programs lack this data or cannot easily obtain it experimentally.
SandboxAQ's structure-generation technology fills that gap. It produces models of both receptor states, including for targets where complete experimental structures are unavailable, giving the platform the foundation it needs to assess how a compound is likely to behave once it binds.
With those models in place, AQState narrows the field. Machine learning rapidly filters large compound libraries–often millions of molecules–down to the most probable binders, eliminating weak candidates before more intensive analysis begins. In retrospective GPCR benchmarking, this filtering step achieved 79% accuracy and 83% specificity. The shortlisted compounds then receive both an affinity ranking and a mechanism-of-action prediction (agonist or antagonist) before any lab work begins.
The approach has demonstrated strong performance across standard Class A GPCRs and, in preliminary benchmarking, on more complex peptide-receptor systems, including the GLP-1 receptor. Those systems are notoriously difficult to model due to molecular size and flexibility; early results suggest AQState handles them well.
SandboxAQ validated the mechanism-of-action approach in collaboration with an industry partner, with findings presented at a major drug discovery conference. That work showed that physics-based calculations could reliably distinguish agonists from antagonists before experimental testing, establishing the concept in a real-world discovery context.
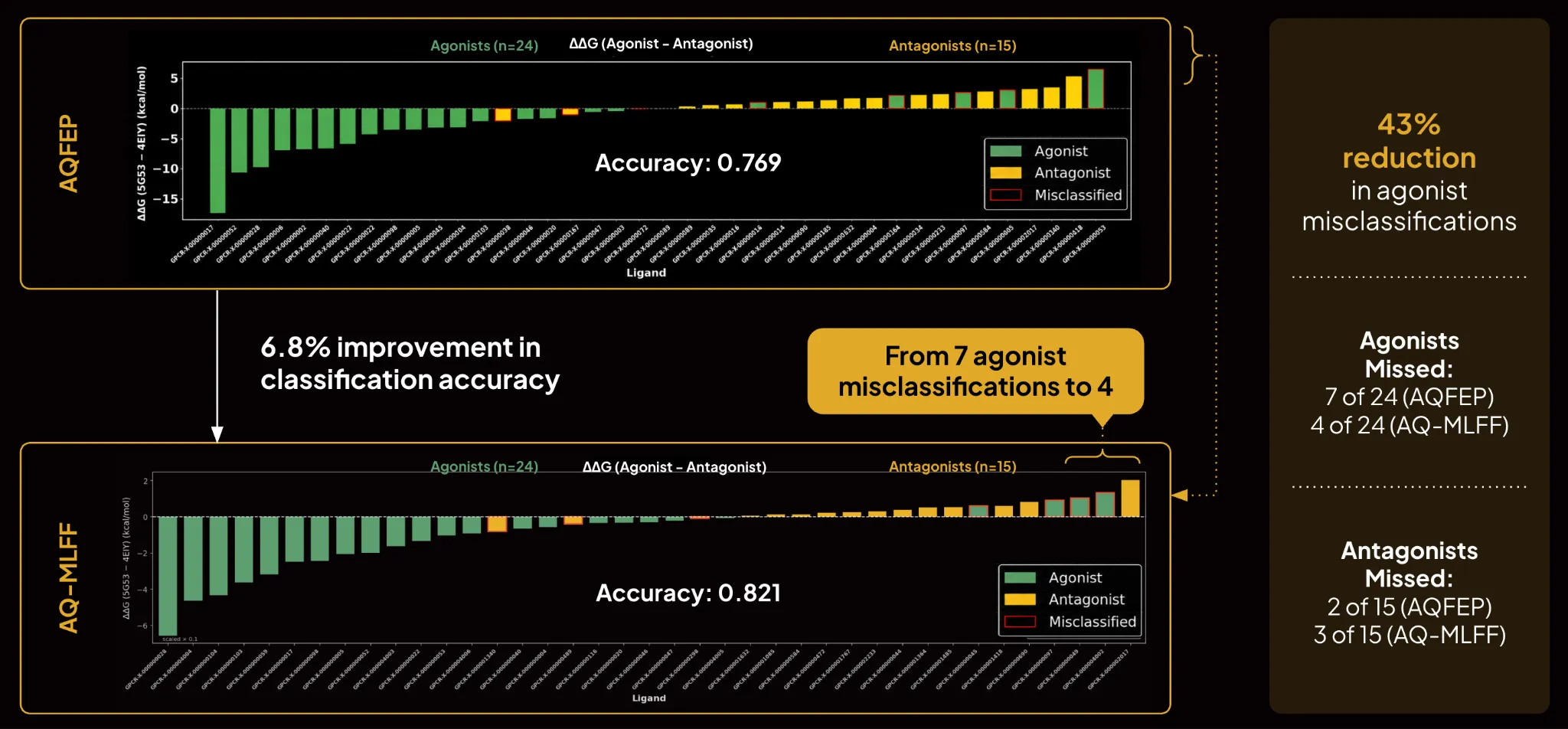
The current generation of the platform improves on those results. In a representative GPCR benchmark, the updated method achieved a 6.8% improvement in overall classification accuracy compared to the previous approach. It missed 4 of 24 agonists and 3 of 15 antagonists, compared with 7 of 24 agonists and 2 of 15 antagonists previously. That is a 43% reduction in agonist misclassifications. For programs where agonism is the target pharmacology, fewer misclassified agonists directly reduces the number of wrong compounds advancing through the funnel.
Binding affinity predictions show comparable or improved accuracy relative to the prior method. Teams gain the mechanism-of-action capability without sacrificing the affinity signal.
AQ-FEP, the previous method used, took approximately 2–3 hours per molecule. The current approach, AQ-MLFF, runs in under one minute per molecule on equivalent hardware (1 T4 GPU), a greater than 100-fold improvement. That difference meaningfully changes what is feasible within a standard project timeline.
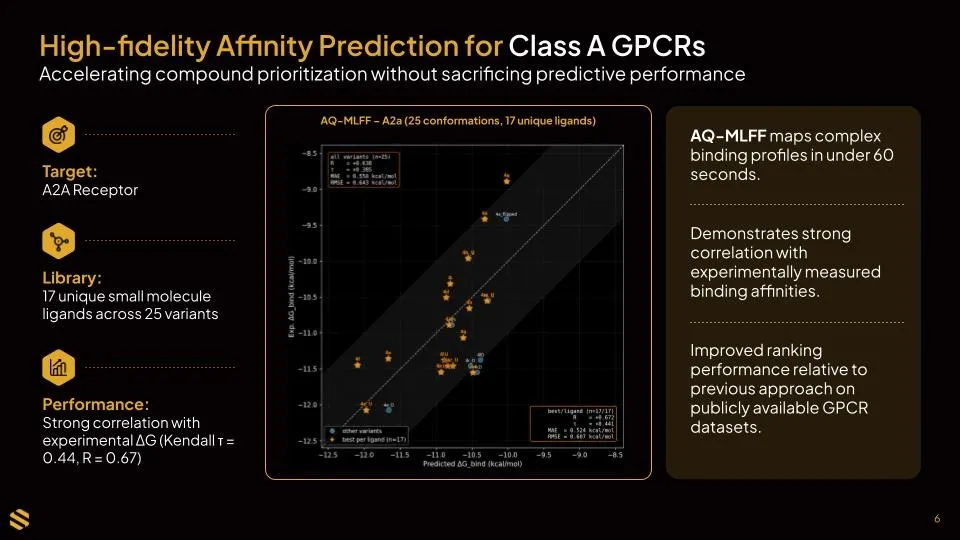
Speed only matters if accuracy holds. Based on retrospective benchmarking, it does: affinity predictions remain at comparable or improved accuracy, and mechanism-of-action classification improves. Teams do not need to choose between rigor and throughput.
For years, the field has treated mechanism-of-action prediction as a downstream problem, resolved experimentally after computational screening rather than predicted alongside it. AQState shifts that logic upstream.
When teams have clarity on both what binds and whether the binding behavior matches the program's therapeutic goal, every subsequent decision improves. Experimental assays focus on a smaller, better-qualified candidate set. Follow-on computational work concentrates where it matters most. By predicting both binding strength and mechanism of action before experimental testing, SandboxAQ helps accelerate GPCR drug discovery while reducing experimental burden and improving candidate prioritization.
In GPCR drug discovery, the bottleneck has rarely been a shortage of binders; it has been a shortage of binders with the right pharmacology. AQState closes that gap before the experiment begins.