In modern pharmacology, the identification and precise tuning of drug-protein interactions are key tools for developing highly efficacious drugs, demanding accurate modelling of structure-activity relationships (SAR).
The advent of deep learning methods for the structure prediction of protein-ligand complexes has opened entirely new avenues for in-silico drug design and for modelling protein-ligand interactions. Trained on large amounts of experimental and high-accuracy synthetic data, models like AlphaFold3, OpenFold3, Boltz2, and Chai are getting progressively better at identifying binding pockets, modelling bound ligands, or identifying critical ligand-residue interactions, and have become a key tool in the toolbox of today’s drug discovery scientists.
A key requirement for drug efficacy is a high binding affinity between the drug and its protein target. To find an initial hit(a compound with high potency that can be further optimized through iterative refinement), labs traditionally have run expensive experimental screens of large compound libraries against a given protein target. For targets with known structure and binding sites, people increasingly use in-silico virtual screening tools like Vina or Gnina to pre-filter compound libraries for likely drug candidates.
Accurate in-silico affinity prediction remains a challenge. The most reliable computational method to date, free energy perturbation (FEP) and its variants, is prohibitively expensive to use in virtual screens. Additionally, FEP requires highly accurate atomistic structures as input, which are often difficult or impossible to obtain experimentally.
Since the spectacular success of AlphaFold, deep learning affinity prediction approaches have drawn increasing interest.
An active direction uses experimental activity measurements to train deep learning models. Chembl, BindingDB, Pubchem or Gostar, for example, are public datasets containing millions of sequence-SMILES pairs with functional or biochemical activity measurements. These, and similar, datasets provide a valuable resource for training ML models, but lack detailed structural information, which is crucial for understanding structure-activity relationships. With current structure prediction models reaching critical thresholds of prediction quality, they can now be used to provide this missing information, and build more accurate affinity prediction models directly on this type of enhanced data, such as our own SAIR dataset.
The recent pre-release of OpenFold3, an entirely open-source reimplementation of AlphaFold3 weights and training data, marks a significant leap forward for the community and is expected to further boost the development of new, powerful deep learning methods for structure and affinity prediction.
SandboxAQ, in collaboration with the OpenFold consortium, is thus today releasing AQAffinity, an open-source affinity prediction model based on OpenFold3 using a Boltz2 architecture. The model is released under the permissive Apache License 2.0 and includes inference and training code, along with assay information from its training data.
AQAffinity was specifically built for predicting small-molecule-to-protein binding affinities. The model predicts affinities directly from sequence-SMILES inputs, without requiring a protein structure.
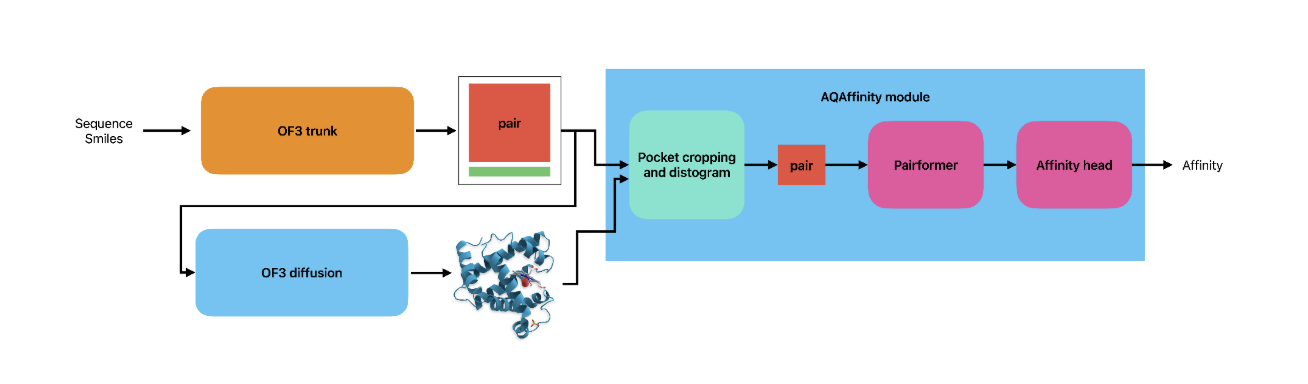
The model architecture follows closely the Boltz2 implementation, and is shown in Fig. 1.
A sequence-SMILES input is passed through the OpenFold3 trunk module to first obtain the single and pair representations of the protein-ligand complex, which are then used to compute the final, folded structure using OpenFold3’s diffusion module.
This structure is then used to crop the pair representation size to contain only tokens (i.e. protein residues and ligand atoms) located around the binding site of the ligand. Finally, the cropped pair representation, enriched with a token-level distogram, is passed through a Pairformer stack and a final affinity prediction head.
The AQAffinity model is run as a standalone application on top of an existing OpenFold3 installation. We provide both inference and training code, so users can train or fine–tune the model on their own data.
The AQAffinity model is trained on a curated dataset from Chembl36, BindingDB and PubChem. Data curation and model training of the affinity head largely follow the procedure outlined in “Boltz2: Towards Accurate and Efficient Binding Affinity Prediction”. Note that the current model doesn’t support a binder classification head at the moment. This feature is currently under development and we plan to add it in a future release.
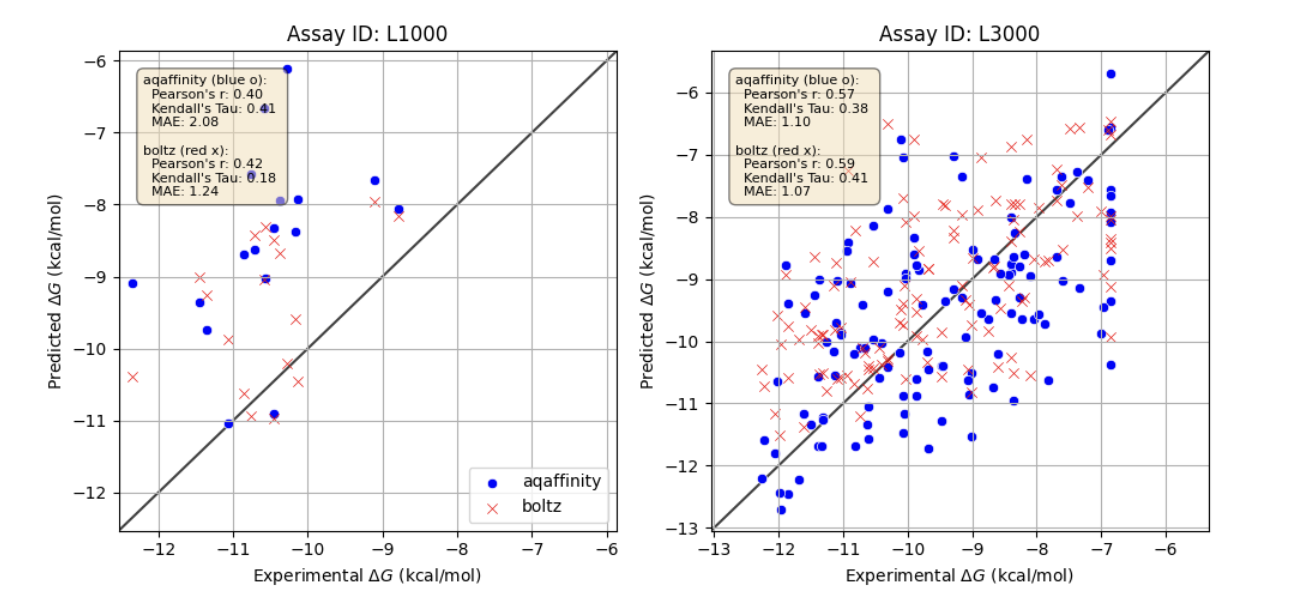
AQAffinity enables accurate, fast prediction of binding affinities of small molecules to proteins without requiring structural information. Below we show retrospective evaluations of AQAffinity on four different assays. Fig. 2 shows predicted activity for the assays L1000 and L3000 from the CASP16 benchmark. For comparison, we also show Boltz2-predicted activities.
While there is room for improvement, performance on CASP16 is already good. However, we note that the L3000 target has high similarity with at least one protein target in the training set (i.e. single point mutation).
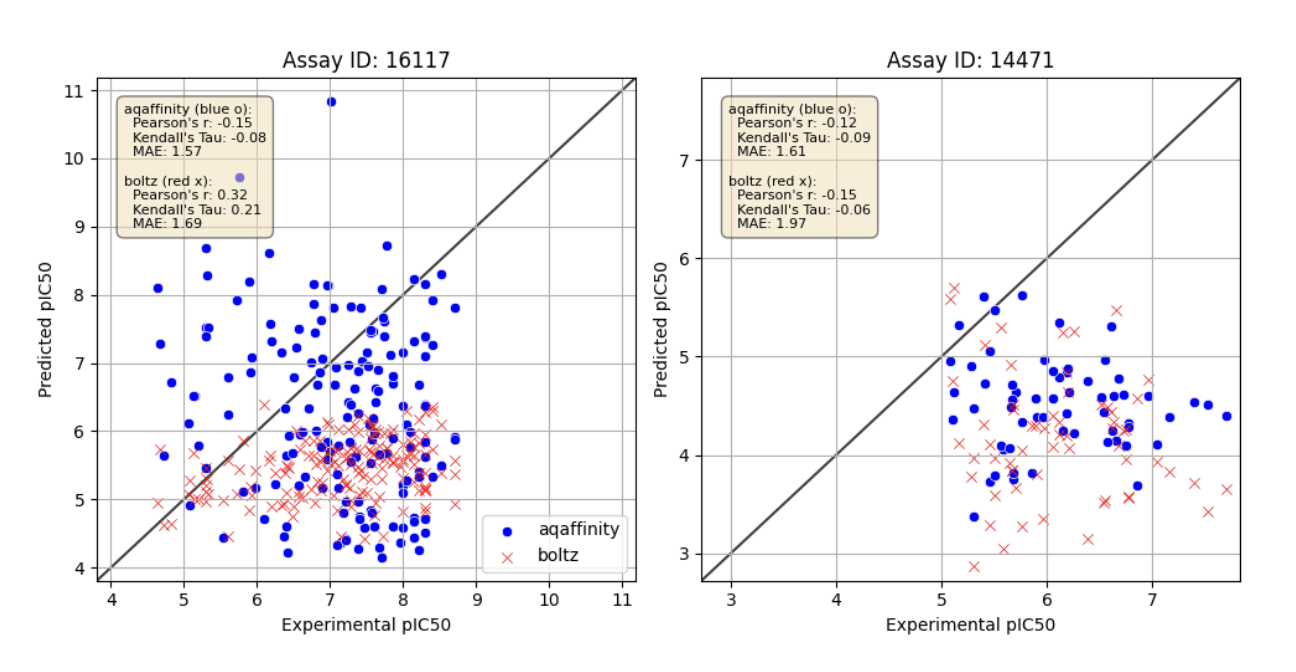
To test out-of-distribution performance of the model, we curated a second test from the GOSTAR assay database. We clustered GOSTAR target sequences according to sequence similarity, and filtered out clusters with sequence similarity larger than 70% to any of the targets in the training data set for Boltz2. From the remaining assays, only those with ligand Tanimoto similarities less than 70% to any of the ligands in the training data assays were kept.
The resulting two assays 16177 and 14471 are shown in Fig. 3.
Model performances for these assays are significantly worse for both Boltz2 and AQAffinity. This illustrates further opportunity to improve generalization, which we will pursue in our later releases.
Reliably predicting drug activities and efficacies in-silico is one of the key challenges in modern drug discovery and development. Accurate protein-ligand structures are a key requirement for many of the most accurate and successful computational approaches to predict structure-activity relationships. Open-source, data-driven co-folding methods like OpenFold3 have the potential to significantly change and improve structure-based affinity and activity prediction, helping scientists to find better drugs earlier and faster. While we’re still at the beginning of this journey, we hope that with the open-source release of AQAffinity under a permissive Apache 2.0 license we can help accelerate this process.