What Are Machine Learning Interatomic Potentials
(MLIPs)?
A machine learning interatomic potential, or MLIP, is an AI model that predicts the energy and forces of a system of atoms. It learns these predictions from large databases of quantum chemistry calculations, then reproduces them at a tiny fraction of the original computational cost. The result is a tool that approaches the accuracy of first-principles physics while running fast enough to screen thousands of candidate structures.
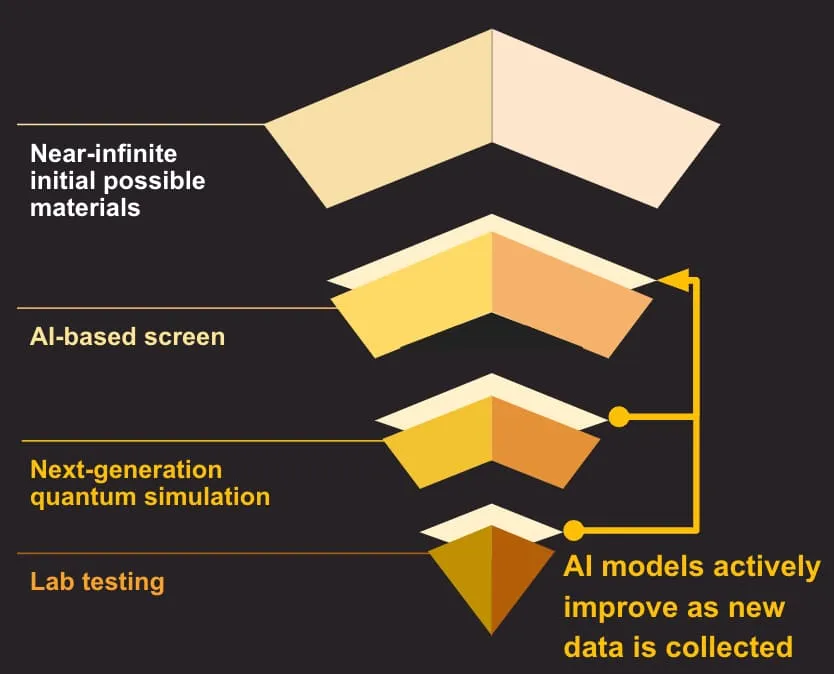
In catalysis, this speed changes what is possible. Designing a better catalyst means understanding how molecules bind and react on a material's surface, and the gold-standard method for that, density functional theory, is too slow to apply across a wide search space. MLIPs let researchers explore that space first in software, reserving expensive calculations and lab work for the most promising candidates.
This guide explains how MLIPs work, why the treatment of magnetism has been a stubborn gap in the field, and how spin-aware datasets such as AQCat25 are extending these models to the catalysts industry actually depends on.
From DFT to machine learning: why speed became the bottleneck
For three decades, density functional theory has been the workhorse of computational catalysis. It links the atomic-scale chemistry of a surface to the reaction rates researchers can measure, and it has guided the design of real catalysts. Its weakness is cost. A single DFT calculation is expensive, which in practice confines the method to relatively simple reaction networks on idealized surfaces of one- or two-element materials.
Machine learning interatomic potentials emerged as a way around that ceiling. Instead of solving the underlying physics for every new structure, an MLIP learns the relationship between atomic arrangements and their energies from a large body of existing DFT calculations. Once trained, it estimates electronic-structure properties at close to quantum accuracy for a small fraction of the compute. That combination of accuracy and speed is what lets teams run physics-based simulation across an entire library of materials rather than a handful.
How a machine learning interatomic potential works
Most modern MLIPs represent a structure as a graph. Each atom is a node, and the connections between nearby atoms carry information about distances and geometry. The model passes information along these connections and predicts two things: the total energy of the system and the force on each atom. Energies tell researchers how stable a configuration is, and forces tell them which way the atoms will move as a structure relaxes toward its lowest-energy state.
The architectures behind these models are built to respect the symmetries of physics, so that rotating or shifting a structure does not change the prediction in ways it should not. This is what allows a single trained model to generalize across many different chemistries and surface geometries. For a broader look at how simulation predicts molecular behavior before anything reaches a lab, see our explainer on AI molecular simulation.
The magnetism gap: why spin matters in catalysis
There is a long-standing limitation hiding inside most large catalysis datasets: they leave out spin. Calculations that account for spin polarization, the magnetic behavior of electrons, are considerably more expensive than calculations that ignore it. When the goal is to generate data at massive scale, spin is often dropped to save time.
That trade-off has a real consequence. Models trained on spin-free data struggle with the earth-abundant transition metals at the heart of industrial chemistry. Iron, cobalt, and nickel show strong spin effects on how molecules bind and how reactions proceed, and these metals drive some of the most important catalytic processes in the world, including ammonia synthesis for fertilizer and Fischer-Tropsch synthesis for fuels. As the field works to replace scarce precious metals with cheaper, more sustainable alternatives, getting magnetism right becomes essential rather than optional. The same challenge runs through the wider field of AI materials discovery.
How spin-aware datasets close the gap: the AQCat25 approach
One way to address the magnetism gap is to build a dataset that treats it directly. AQCat25 is a worked example of that approach. It is a collection of 13.5 million density functional theory single-point calculations spanning roughly 47,000 catalyst systems, designed specifically for cases where spin polarization and higher fidelity matter.
Three choices make it suited to industrially relevant catalysis. It enables spin polarization for twelve magnetic elements, so metals like iron, cobalt, and nickel are described correctly. It raises the plane-wave cutoff, a key accuracy setting, to 500 eV for more reliable energies, especially on materials containing non-metals. And it adds six elements never before included in catalysis-focused datasets, barium, cerium, fluorine, lithium, lanthanum, and magnesium, opening the door to new candidate catalysts. Together these features give models reliable reaction and barrier energies across a broader slice of chemistry. The dataset and the resulting models are described in detail on the AQCat25 page, and the principles behind translating this kind of data into design decisions are covered in our guide to computational materials engineering.
Training a model across mixed physics: catastrophic forgetting and joint training
Building a spin-aware dataset is only half the problem. The other half is teaching a model to use it without losing what it already knows. When researchers take a capable general-purpose model and fine-tune it only on the new spin-aware data, the model improves on the new material but forgets much of its earlier knowledge, a failure mode known as catastrophic forgetting.
The fix is to train on both sources at once rather than in sequence. Joint training preserves broad, general performance while still gaining accuracy on the new, harder cases. This introduces a subtler challenge, because the combined data now mixes different levels of fidelity and different physics, with some calculations including spin and others not. The solution is to tell the model which is which. By feeding it simple context about each structure's spin treatment and fidelity, the model learns to keep the two regimes straight instead of blurring them together, which improves accuracy further. This conditioning technique is what allows one model to serve many physical situations at once.
What this enables for catalyst discovery
The practical test of any of these models is whether they can find the most stable way a molecule sits on a surface, the quantity researchers use to rank candidate catalysts. On this task, accuracy climbs steadily as the training strategy improves. Across the model variants studied with AQCat25, the share of predictions landing within a tight tolerance of the reference value rose from roughly 48 percent for a baseline model to about 64 percent after direct tuning and to nearly 70 percent for the best jointly trained model.
That progression matters because it maps onto real industrial questions, screening catalysts for cleaner ammonia production, for fuel synthesis, and for the broader set of reactions that depend on earth-abundant metals. Teams can rank candidates in software before committing to costly synthesis and testing, which is the core promise of AI-accelerated catalyst and materials discovery. It is also part of a wider shift toward AI for the physical world, where models grounded in physics tackle energy and industrial problems directly.
Frequently asked questions
What is a machine learning interatomic potential?
It is an AI model that predicts the energy and forces of a group of atoms after learning from large databases of quantum chemistry calculations. Once trained, it delivers near-quantum accuracy at a small fraction of the computational cost, which makes large-scale screening of materials practical.
How accurate are MLIPs compared to DFT?
Well-trained MLIPs approach the accuracy of the density functional theory data they learn from, while running orders of magnitude faster. Accuracy depends heavily on the quality and coverage of the training data, which is why dataset design is such an active area of research.
Why is spin polarization important for catalysis modeling?
Many industrially important catalysts rely on magnetic metals such as iron, cobalt, and nickel, whose spin behavior strongly affects how molecules bind and react. Models trained on data that ignores spin tend to describe these materials poorly, so capturing spin is essential for realistic predictions.
What is the AQCat25 dataset?
AQCat25 is a large-scale, publicly available dataset of 13.5 million DFT single-point calculations covering about 47,000 catalyst systems. It is notable for explicitly including spin polarization, using higher-fidelity settings, and adding six elements not found in earlier catalysis datasets.
Are MLIPs and the AQCat25 models publicly available?
The AQCat25 dataset and its associated models are released publicly on Hugging Face under a Creative Commons non-commercial license, so academic and research teams can build on them directly.
Want to apply spin-aware machine learning to your own catalyst and materials challenges? Contact our team to start the conversation.
Read the full peer-reviewed research: AQCat25 in npj Computational Materials.